Last year, Zhen Liu et al., from the Institute of Neuroscience of the Chinese Academy of Sciences cloned Macaque monkeys (Macaca fascicularis), by somatic cell nuclear transfer (SCNT), the same cloning technique used to produce Dolly the sheep.
Cloning of animals by SCNT has been achieved in 23 mammalian species, the first of which was Dolly the sheep in 1996. As non-human primates are closely related to humans they are the ideal choice for animal models when studying humans diseases and developing therapeutics. Compared to rodents, the breeding of non-human primates takes a long time, therefore inbreeding approaches for generating animal models with genetic uniformity are not desirable. It is for this reason that SCNT has been explored.
The technique of SCNT involves removing the nucleus from an oocyte (egg cell) and implanting a donor nucleus into it from a somatic (body) cell. The nucleus fuses with the cell and the resulting viable embryo can be implanted into a surrogate mother.

(Modified from: en: converted to SVG by Belkorin, modified and translated by Wikibob [CC BY-SA 3.0 (http://creativecommons.org/licenses/by-sa/3.0/)])
Unlike the previous attempts at generating cloned mammals, including non-human primates, this study was the first to successfully generate two monkeys using fetal fibroblast cells, instead of embryonic fibroblast cells- these are a type of cell that can be found in connective tissue. Previous studies using the same type of cells resulted in pregnancies lasting up to 80 days. This study highlights that failure in the generation of successful clones prior to this work was due to poor reprogramming of the nuclei. This meant that when they were transferred to the somatic cells the nuclei couldn’t support embryonic development. Before implanting the embryos into the surrogate, the group added two molecules to the cell growth media which disrupted proteins that normally prevent transcription of genes, these were messenger RNA and trichostatin A. This allowed for reprogramming of the nucleus for development, activating transcription of genes vital for various stages of embryonic development instead of somatic functioning.
They transferred 79 embryos into surrogate mother monkeys. Of the resulting 6 pregnancies, two produced living animals. Although the success rate was low, it is a major improvement upon the creation of Dolly which required 277 attempts, producing only one living lamb. The success of this work paves the way for developing better animal models to understand both the pathology and progression of disease with the goal of producing therapeutics.
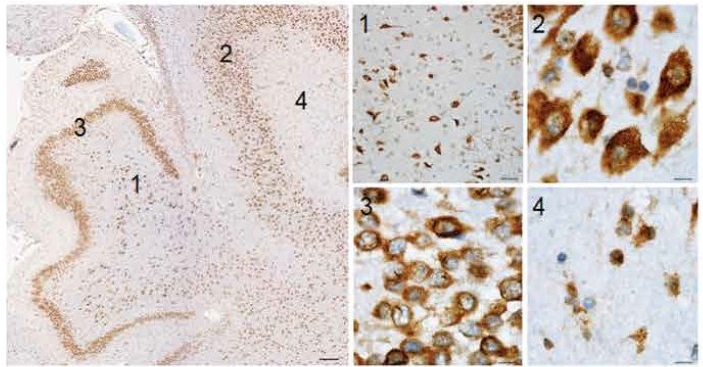
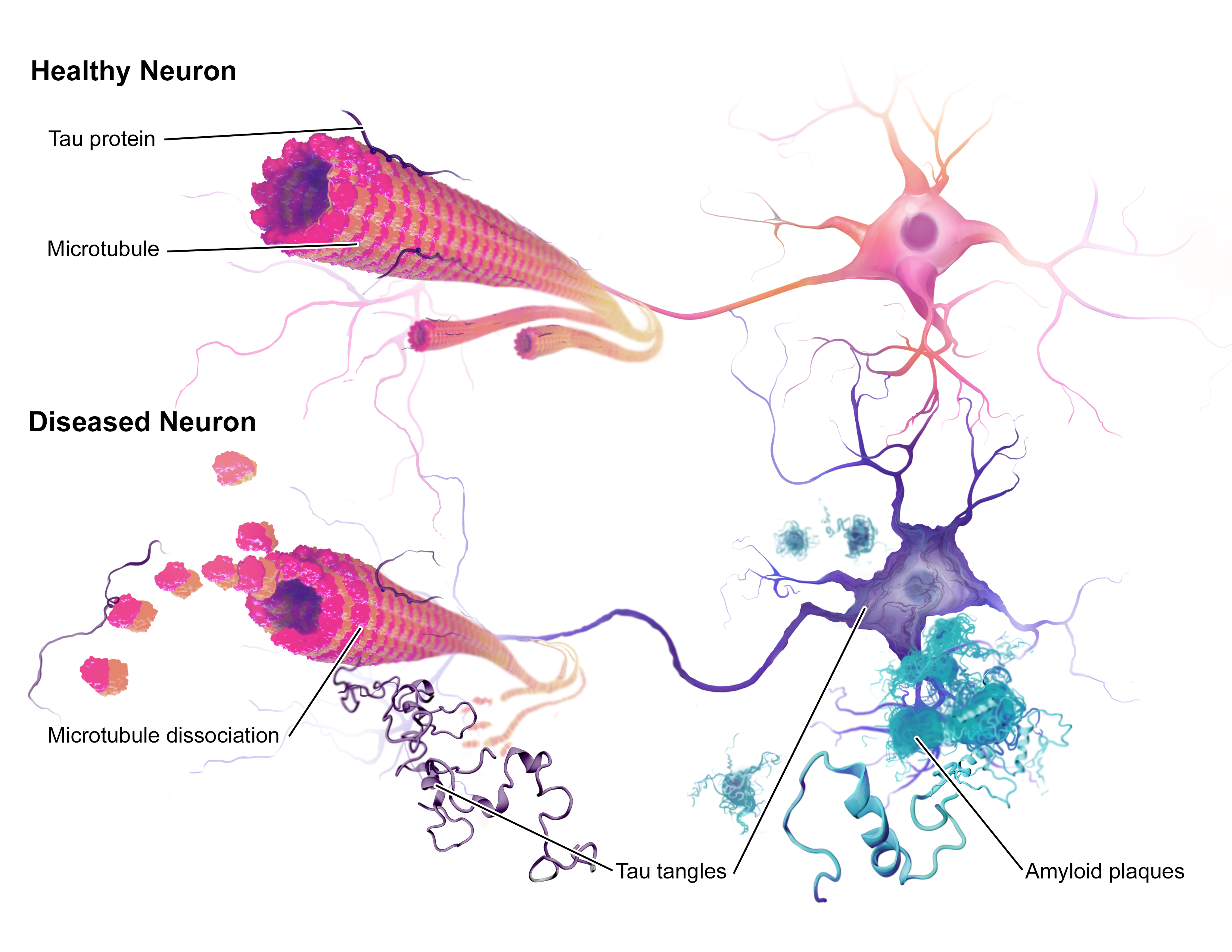
This is certainly an admirable concept given the difficulties faced thus far when trying to mimic complex diseases like Alzheimer’s in mice. Therapies that have successfully treated Alzheimer’s-like symptoms in mice have failed when trialled in humans. One reason for this could be that the mouse model of Alzheimer’s is not a close enough replica of the human disease, due to the lack of similarity between species. With non-human primates being closely related to humans, they may prove to be a better model for Alzheimer’s and other human diseases.
After this study was published it sparked discussions as to what this work means for the future of genetic cloning. The question of whether human cloning is possible was raised, to which one of the lead authors, Mu-ming Poo, responded,
Technically speaking one can clone humans…But we’re not going to do it.
The future of this work will not only likely be involved in therapeutics but in gene editing. This will allow for gene knock-out or knock-in monkey embryos with defined genetic defects for the study of a variety of diseases, allowing us to gain a better understanding of their mechanisms.
Personally, I am not sure how I feel with regards to the success of this work. On the one hand it is an astonishing achievement to have produced living non-human primate clones, given the history of unsuccessful attempts since Dolly the sheep was produced. Moreover, because of the close relation between non-human primates and ourselves with them having advanced minds as well as complex social networks, they are likely to show us more about diseases than mice ever could, particularly those affecting the brain. However, the cost of housing primates is expensive and a significant number of them would need to be produced for them to be a useful method of study. In the UK, due to the ethical restrictions surrounding primate testing, they are only used when there are no other means to address questions of importance. Therefore, current mouse models would still be sufficient in most cases, meaning that genetically uniform primates would be unnecessary. Moreover, being vegan, the treatment of animals is paramount to me. Entering into the world of research, I recognise the importance of animal models for understanding the complexity of diseases in whole organisms but I hope that one day we will be able to move away from them completely.
Drop me a comment down below about your thoughts on genetic cloning and the use of animals in research 🙂